By Gaby L. Longsworth, Alex Wang, and Dennies Varughese
Attorneys with Sterne Kessler examine a pathway for getting drugs on the market that may avoid the expense of a new drug application and the pitfalls of a crowded generic market: the 505(b)(2) new drug application. The authors say that in a global environment where the cost of developing a branded drug is skyrocketing and price pressures loom large, a drug manufacturer would be wise to incorporate 505(b)(2)s in its pipeline and portfolio strategy.
Introduction
Drug manufacturers seeking to introduce new branded medicines must first submit, and obtain approval from the U.S. Food and Drug Administration (“FDA”), a New Drug Application (“NDA”), codified under FDCA § 505(b)(1). (21 U.S.C. Chapter 9 – Federal Food, Drug, And Cosmetic Act.) The NDA is an extensive dossier containing preclinical and clinical studies establishing the safety and efficacy of the proposed drug. An NDA is cost prohibitive for many manufacturers, and could range from the hundreds of millions to approaching a billion U.S. dollars.
On the other end of the spectrum, manufacturers seeking to introduce low-cost generic versions of branded drugs need submit only an Abbreviated New Drug Application or “ANDA,” codified under FDCA § 505(j). An ANDA is drastically less expensive than an NDA, and need only demonstrate “bioequivalence” to the corresponding branded product, rather than full studies showing safety and efficacy. But in turn, the ANDA must meet the FDA’s “sameness” standard with the brand product—i.e., the proposed generic must be identical with respect to active ingredient, salt form, dosage strength, dosage form, and route of administration. And for some dosage forms, such as ophthalmics, the ANDA must be virtually identical across the board, including with respect to most inert excipients, leaving little room to design around patents covering the branded drug.
In the thirty-four years since passage of the Hatch-Waxman Act (The Drug Price Competition and Patent Term Restoration Act of 1984 – Public law 98-417)—widely credited for essentially creating the generic drug industry—the number of generic competitors in the marketplace has steadily grown, substantially reducing profits for the generic industry as a whole and reducing the incentives for generic companies to risk challenging patents to enter the market early. Likewise, the stringent “sameness” requirements for ANDAs limits the ability for generic manufacturers to design around patents covering branded drugs, rendering it more difficult to overcome patent protection, raising the costs of litigation, thereby further reducing profits.
Background on 505(b)(2) Applications
Faced with these challenges, more generic companies are turning to a second, and historically overlooked (February 2016 source; see also October 2009, source,) drug approval pathway—the 505(b)(2) NDA or “Paper NDA”—which is a hybrid pathway that shares characteristics of both an ANDA (505(j)) and a full NDA (505(b)(1)). For example, like an ANDA, a 505(b)(2) application is abbreviated to some extent, and references (or “piggybacks” onto) an existing approved branded drug, relying on the safety and effectiveness data of the reference-listed-drug (“RLD”). That way, the 505(b)(2) applicant may avoid much of the costly and extensive preclinical and clinical safety and efficacy testing (21 USC §355(b)(2).)
But, unlike an ANDA, the 505(b)(2) pathway does not have the same “sameness” requirement, and allows for modifications to the proposed product to deviate from the RLD in many aspects, including the salt form, dosage form, route of administration, strength, new combination product, modified active ingredient, new indications for previously approved drugs, or an over-the-counter switch. (April 2010, source.) And the 505(b)(2) is more expensive than an ANDA (but vastly cheaper than a full NDA) in that the applicant typically must conduct some clinical studies to bridge the difference between its proposed product and the reference brand product. An approved product under the 505(b)(2) pathway can be designated either as a true generic that is AB-rated, a non-AB rated or “branded” generic, or a stand-alone branded drug, depending on the extent and nature of the deviation from the RLD. (November 2008, source.)
This article explores the advantages and limitations of the 505(b)(2) pathway compared to the full 505(b)(1) NDA and ANDA, specifically examining four case studies providing real-world insight into the usefulness of 505(b)(2)s. (FDA October 2017, source.)
Table I below illustrates the similarities and differences between a 505(b)(1) (NDA), a 505(b)(2) and a 505(j) (ANDA) application.
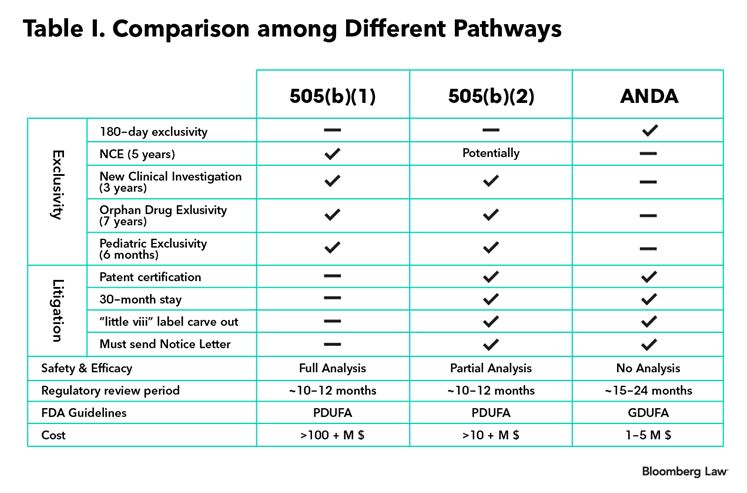
In short, the 505(b)(2) pathway presents an important strategic tool for generic drug manufacturers to distinguish themselves and derive substantial value in an increasingly competitive generic marketplace.
Examples of 505(b)(2) Approved Products
Austedo—Obtaining NCE Exclusivity for a Follow-On Product
Recently, Austedo—a deuterated version of tetrabenazine—was approved under Section 505(b)(2). (NDA 208082, April 2017, source.) A deuterated drug has one or more hydrogen atoms replaced by deuterium – a heavier stable isotope of hydrogen. Due to the kinetic isotope effect, deuterium version drugs might have significantly lower rates of metabolism, and hence longer half-life. The reference-listed-drug, non-deuterated tetrabenazine (sold under the tradename Xenazine), was previously approved by the FDA in 2008. Yet, Austedo was able to earn New Chemical Entity (“NCE”) status because the structural difference between Austedo and the reference-listed-drug—two groups of hydrogens replaced with deuteriums (Figure 1)—rendered the two drugs sufficiently distinct under the FDA’s regulations.
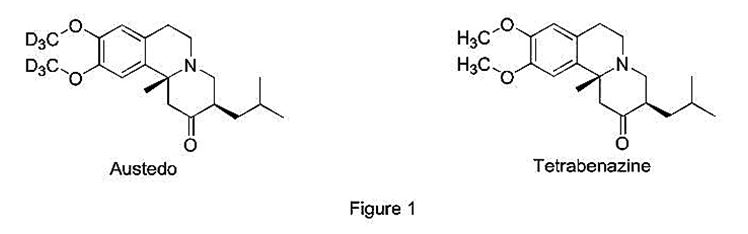
Because the FDA classified Austedo as a “new” drug, it was eligible for 5-year market exclusivity awarded to NCE drugs, which typically is reserved for drugs approved under the full 505(b)(1) NDA. The 5-year exclusivity is a major windfall for a 505(b)(2) applicant, when considering that the highest incentive for a typical ANDA applicant who is the First-to-File is only 180 days of generic exclusivity. (21 U.S.C. § 355(j)(5)(B)(iv)(I).) To earn NCE status and the corresponding 5-year exclusivity, the FDA must conclude that the proposed 505(b)(2) drug is not the “same drug” as the reference-listed-drug. (July 2015, source.) This determination is generally based on a “structure-based approach,” (Id.) where the FDA looks for structural differences that involve “non-ester covalent bonds.” (Id.) In this instance, the presence of deuteriums in Austedo results in a structural difference that involves a non-ester covalent bond, thus rendering Austedo not the “same drug” as tetrabenazine under the FDA’s standard, even though the difference between the two drugs is only one neutron.
Amlodipine Maleate/Besylate—Allowing subsequent generic applicant to circumvent a First-to-File Generic Applicant’s 180-day Exclusivity
Another strategic use of a 505(b)(2) is to circumvent a first ANDA filer’s 180-day exclusivity. For example, Pfizer received approval for its branded blockbuster hypertension medication Norvasc® (amlodipine besylate) in 1992. A first generic applicant submitted an ANDA containing a Paragraph IV certification for this product, and thus earned eligibility for 180-day exclusivity that could potentially block all other subsequent ANDA filers from the market indefinitely. As an ANDA, the first-filers’ proposed product had to be the same “besylate” salt as the reference Norvasc® product.
Another generic company, Dr. Reddy’s Labs, sought to market its own generic version of Norvasc®. (Nov. 4, 2003, source.) However, the first-filer’s 180-day exclusivity would block Dr. Reddy’s from launching a generic version of amlodipine besylate at the same time as the first-filer, leaving Dr. Reddy’s at a competitive disadvantage. Dr. Reddy’s then filed a 505(b)(2) to market amlodipine maleate, a different salt form of the branded drug. Because this product would be approved as a 505(b)(2) NDA, and not as an ANDA, it was not subject to or blocked by the first-filer’s 180-day exclusivity. Pfizer/Torpharma challenged Dr. Reddy’s 505(b)(2) application, arguing that a “suitability petition” was more appropriate for the switch to the maleate salt. (FDA Docket No. 2002p-0447, p27-28, source. (both Pfizer and Torpharma argued that Dr. Reddy’s 505(b)(2) was not proper. Torpharma focused on the question of whether a “suitability petition” was more appropriate for the switch to the maleate salt. Pfizer argued whether a 505(b)(2) should be same as a suitability petition.)) A suitability petition is a mechanism where, even though the generic application deviates from the FDA’s “sameness requirement” for characteristics such as salt form, the FDA may nevertheless determine that the product is still “suitable” to be filed as an ANDA, and thus without the need for any additional bridging safety or efficacy studies. (21 U.S.C. § 355(j)(2)(C).)
Torpharma, of course, knew that if Dr. Reddy’s had filed a suitability petition and received FDA-approval as an ANDA, then Dr. Reddy’s would be indefinitely blocked from launching a generic because it would be subject to the first-filer’s 180-day exclusivity. The FDA sided with Dr. Reddy’s and allowed the 505(b)(2) application to proceed through review, reasoning that the use of a suitability petition for a change in active ingredient is allowed only where “one active ingredient is substituted for one of the active ingredients in a listed combination drug,” (Id., at 27) a circumstance not present in this case because amlodipine is the only active ingredient. By electing to file a 505(b)(2) in lieu of an ANDA, Dr. Reddy’s was able to circumvent the first-filer’s generic 180-day exclusivity, thereby gaining a potential market advantage.
Mitigare—505(b)(2) Provides Flexibility to Choose RLD, thereby avoiding 30-month stay of ANDA litigation
The branded drug here was Colcrys (colchicine), owned by Takeda and approved in 2009. However, the active ingredient—Colchicine—was a “desi drug,” had been on the market for decades beforehand, and was also the active ingredient in another combination product called Col-Probenecid (colchicine + probenecid) to treat gout. (Desi drugs are a class of drugs which had existed in the market prior to 1962 and were found safe and effective through a program called Drug Efficacy Study Implementation (DESI).) Various generic manufacturers submitted ANDAs for generic versions of Colcrys containing Paragraph-IV certifications challenging its Orange-Book-listed patents, and sent the required “Notice Letter” with the Paragraph IV certifications to Takeda. In response, and as expected, Takeda brought suit within the 45-day statutory period after receiving the notice, securing the 30-month stay of FDA approval of each of those ANDAs while the district court case was pending.
However, Hikma, another generic applicant, submitted a 505(b)(2) application for a generic colchicine, referencing Col-Probenecid as the RLD. Notably, Col-Probenecid was no longer covered by any patents, (July 16, 2016, source) so Hikma did not have to certify to any Orange Book patents. (Takeda v. Burwell, 691 F. App’x. 634 (D.C. Cir. 2016).) Because Hikma did not reference Takeda’s Colcrys, it did not have to certify to Takeda’s patents or provide any “Notice Letter” to Takeda. Thus, Takeda did not even learn of Hikma’s filing until Hikma received final approval for its colchicine product (sold under the name Mitigare) and was ready to launch. And even though Takeda immediately brought an infringement action upon learning of Mitigare’s approval, it was not the type of suit that triggered the same 30-month stay of approval to which the ANDA applicants were subject. Moreover, as a 505(b)(2) NDA, Mitigare could receive FDA approval approximately 10 months after filing, whereas the other ANDA filers were all still subject to the 30-month stay as the district court case was ongoing.
Takeda challenged the FDA’s approval of Mitigare in the District Court for the District of Columbia and argued that Hikma should have (1) referenced Colcrys as the RLD and (2) submitted Paragraph IV certifications to the Colcrys patents with the corresponding Notice Letter. (Takeda v. Burwell, 78 F.Supp.3d 65 (D.C. 2015).) The district court sided with the FDA by finding no basis in law or fact for Takeda’s request that the FDA was legally required to force Hikma to reference Colcrys and to certify to the Colcrys patents. (Id., at 85.) Takeda appealed to the Circuit Court. The Court did not answer this question directly, but instead dismissed the appeal by concluding that the issue was moot because Takeda could not prove that Hikma infringed Takeda’s Colcrys patents in a separate parallel patent infringement action pending in the District of Delaware. (Takeda v. Burwell, at 637.)
The Court explained even if Hikma should have certified to Takeda’s patents, that decision would at most entitle Takeda to a stay of FDA’s approval of Mitigare pending a district court decision on the patent infringement suit. (Id.) But because there had already been a district court judgment on the patent infringement suit, Takeda would not receive any stay of FDA’s approval of Mitigare. (Id.) Without the possibility of such a stay, Takeda’s claims about Hikma’s failure to certify to the Colcrys patents were academic and moot. (Id.) Although it is not entirely clear how far a 505(b)(2) applicant can go in choosing a listed drug as its reference, (July 16, 2016, source) the Hikma case illustrates the benefit of using the 505(b)(2) pathway to avoid the 30 month stay, avoid protracted paragraph IV litigation, obtain early approval, with the net effect being the ability to bring the drug to market more rapidly and, at least in this instance, to a de facto exclusive generic market.
Yosprala (aspirin-omeprazole delayed release tablets)—a 505(b)(2) as a mechanism to introduce a new branded product
Finding new therapeutic uses for known drugs, developing different formulations for the same drug, or creating new combinations of at least two drugs previously used as separate drug products each present opportunities to penetrate new markets and derive substantial revenue. (February 2014, source.) A 505(b)(2) serves to achieve any of these objectives. And when using a 505(b)(2) to introduce a new branded product employing these strategies, the applicant can also obtain its own patent protection for listing in the Orange Book to block competitors.
Take for example the case of Yosprala, which is a new combination of two existing generic drugs: aspirin, an anti-platelet agent, and omeprazole, a proton pump inhibitor. While aspirin and omeprazole have commonly been prescribed together, Yosprala is not a therapeutic equivalent of, and cannot be interchanged with, the individual component of aspirin and omeprazole. To bridge the difference, Yosprala was studied in two randomized clinical trials for safety and efficacy. (September 21, 2017, http://www.genericsand505b2.com/wp-content/uploads/2017/09/Shivanand_9.21.17.pdf.) By relying on these clinical trials, previous studies, and some pharmakinetic and pharmacodynamic (PK/PD) studies, the FDA approved Yosprala in 2016. (NDA Approval Letter – NDA 20513, September 2016, http://www.genericsand505b2.com.) And the applicant for Yosprala obtained a patent to cover this combination, U.S. Patent No. 9,539,214, which is listed in the Orange Book and will not expire until 2033. (Patent and Exclusivity for: N205103, source.) This means that any generic applicant for Yosprala must certify as to this patent, which will trigger a lawsuit and at least 30 months of additional exclusivity or longer if the applicant prevails in any ensuing patent litigation. Although Yosprala’s exact annual sales numbers have not been reported, Aralez, the applicant holder of Yosprala, has stated that Yosprala sales are an important driving force for the company’s growth. (March 2017, source.)
* * *
The above four cases illustrate just some of the ways in which a 505(b)(2) strategy can help manufacturers gain market exclusivity, circumvent a first ANDA filer’s 180 day exclusivity, avoid the 30 month stay, and repurpose existing drugs into new branded products that can be sold directly or through an asset transfer, license, or other monetizing opportunity.
Table II below shows the revenue stream generated by certain other notable drugs approved under the 505(b)(2) pathway. (See, 2017, source.)
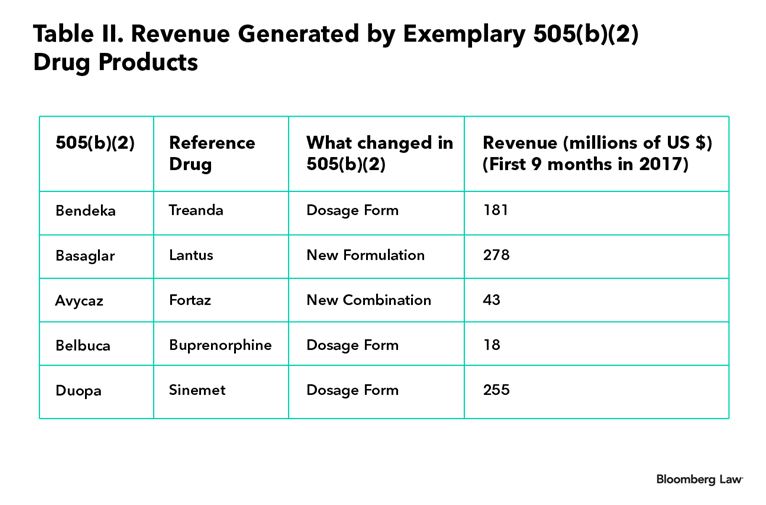
As Table II shows, some 505(b)(2) drugs have generated hundreds of millions of dollars in revenue in just nine months, which is comparable to many drugs approved under the 505(b)(1) pathway. Considering that 505(b)(2)s are less expensive and carry lower risks of failure, this approach may provide attractive returns on investment for a drug manufacturer. In summary, in a global environment where the cost of developing a branded drug is skyrocketing and price pressures loom large, a drug manufacturer would be wise to incorporate 505(b)(2)s in its pipeline and portfolio strategy.
Gaby L Longsworth, Ph.D. and Dennies Varughese, Pharm.D. are directors at Sterne, Kessler, Goldstein & Fox in Washington, D.C. Alex Wang is a law clerk at the firm. Gaby Longsworth is on the advisory board for Bloomberg Law.
Related Industries
Related Services
Receive insights from the most respected practitioners of IP law, straight to your inbox.
Subscribe for Updates